La enfermedad de Gaucher (EG), fue descrita por primera vez por el médico francés Philippe Charles Ernest Gaucher, quien nació el 26 de julio de 1854. Gaucher se refirió a esta enfermedad en su tesis de doctorado titulada: “De l’épithélioma primitif de la râte; hypertrophie de la râte sans leucémie” (Epitelioma primaria del bazo: hipertrofia idiopática del bazo sin leucemia); en la cual describe un aumento significativo en el tamaño del bazo en una paciente de 32 años que había fallecido.1
Sin embargo, en 1924, el médico alemán Hans Lieb logró separar un compuesto graso del bazo de personas con EG, el cuál fue identificado diez años después por el doctor Henriette Aghion como glucocerebrósido. Finalmente, en 1965, el médico estadounidense Roscoe O. Brady y sus compañeros descubrieron que la acumulación de este componente graso ocurre por una deficiencia de la enzima hidrolasa ácida llamada B-glucosidasa o glucocerebrosidasa. Lo anterior, brindó las bases para poder desarrollar terapias de reemplazo de esta enzima para pacientes con EG.1
El descubrimiento del doctor Brady explicaba que la deficiencia de la enzima glucocerebrosidasa, responsable de descomponer al glucocerebrósido en glucosa y ceramida; causa la acumulación de glucocerebrósidos. Este último, forma agregados fibrilares que se almacenan en el citoplasma de los macrófagos presentando una apariencia de “papel de seda arrugado” como se observa en la siguiente imagen.2
Imagen 1. Aspecto del citoplasma de las células de Gaucher.2
Estas células, también conocidas como células Gaucher, son capaces de infiltrarse en distintos órganos (principalmente el bazo, hígado y médula ósea); siendo responsables de la mayoría de síntomas de la enfermedad. 2
Fisiopatología: La enfermedad de Gaucher se agrupa dentro de las patologías por depósito lisosomal, las cuales incluyen más de 40 entidades genéticas consideradas enfermedades huérfanas al afectar a menos de 10.000 personas mundialmente. Ocurren por la deficiencia de una o más enzimas responsables de degradar lípidos u otras moléculas, lo cual bloquea la ruta catabólica con el acúmulo consecuente de productos metabólicos intermedios.3
Las enfermedades por depósito lisosomal son progresivas, mortales y no tienen cura; sin embargo, las Terapias de Reemplazo Enzimático (TRE) constituyen una alternativa terapéutica que consiste en la administración parenteral de proteínas recombinantes que contrarrestan la insuficiencia enzimática. 3
A continuación, se muestra la clasificación de las enfermedades por depósito lisosomal. La enfermedad de Gaucher pertenece a la Esfingolipidosis, conjunto de enfermedades hereditarias que involucran el almacenamiento anormal de esfingolípidos.
La enfermedad de Gaucher es un trastorno genético autosómico recesivo que cursa con la mutación en el gen GBA1 ubicado en el cromosoma 1 (1q21); esta alteración conlleva a una deficiencia de la enzima lisosómica glucocerebrosidasa por lo que ocurre una acumulación de sustratos glucolípidos, en este caso los glucocerebrósidos en macrófagos; transformándose en células Gaucher.4
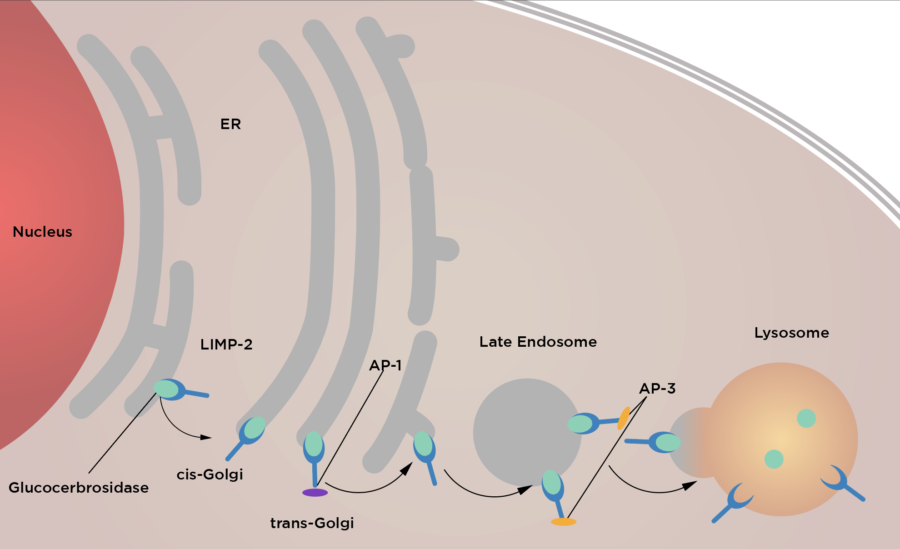
Otra de las causas que conlleva a la acumulación de glucocerebrósidos es la deficiencia de la proteína integral de membrana lisosómica tipo 2 (LIMP-2); en una persona sin EG, LIMP-2 o SCARB2 se une a la B-glucosidasa con la ayuda de la proteína progranulina, y es transportada por el retículo endoplasmático hacia el interior del lisosoma; sin embargo, en un paciente con EG esto no ocurre ya que una anomalía en el ácido aspártico 399, isoleucina 402 o isoleucina 403; interrumpe la asociación de LIMP-2 y la enzima por lo que la enzima glucocerebrosidasa no cumple su papel de degradar los glucocerebrósidos en glucosa y ceramida. 4
Imagen 2. Asistencia de LIMP-2 en el tráfico de GCase desde el RE hasta el lisosoma.5
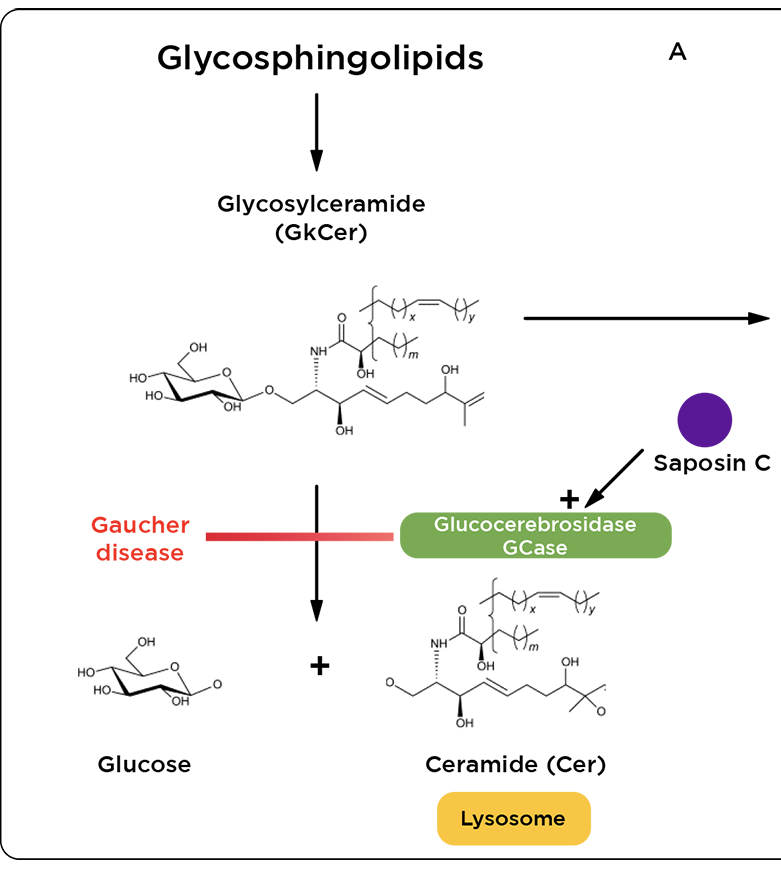
Por otro lado, aunque menos frecuente, puede ocurrir una deficiencia del activador de la glucocerebrosidasa: la Saposina C, por la mutación en el gen PSAP. 5
Imagen 3. Hidrólisis de glucosilceremida (GlcCer) por glucocerebrosidasa (GCase)5
González, E., Aguilar, M., Álvarez, J., García, P. (2010). Enfermedad de Gaucher y su manejo clínico en el paciente pediátrico. Obtenido el 09 de septiembre del 2021, de https://scielo.isciii.es/pdf/albacete/v3n2/especial2.pdf
Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, Levade T, Astudillo L, Serratrice J, Brassier A, Rose C, Billette de Villemeur T, Berger MG. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int J Mol Sci. 2017 Feb 17;18(2):441. doi: 10.3390/ijms18020441. PMID: 28218669; PMCID: PMC5343975.
Gonzalez A, Valeiras M, Sidransky E, Tayebi N. Lysosomal integral membrane protein-2: a new player in lysosome-related pathology. Mol Genet Metab. 2014 Feb;111(2):84-91. doi: 10.1016/j.ymgme.2013.12.005. Epub 2013 Dec 11. PMID: 24389070; PMCID: PMC3924958.
¡Haz clic para calificar esta publicación!
Debes registrarte para votar
No Comments
Lo sentimos, el formulario de comentarios está cerrado en este momento.
Usamos cookies en nuestro sitio web para brindarle la experiencia más relevante recordando sus preferencias y visitas repetidas. Al hacer clic en "Aceptar", acepta el uso de TODAS las cookies.
El acceso a este Sitio Web puede implicar la utilización de cookies. Las cookies son pequeñas cantidades de información que se almacenan en el navegador utilizado por cada Usuario —en los distintos dispositivos que pueda utilizar para navegar— para que el servidor recuerde cierta información que posteriormente y únicamente el servidor que la implementó leerá. Las cookies facilitan la navegación, la hacen más amigable, y no dañan el dispositivo de navegación.
Las cookies son procedimientos automáticos de recogida de información relativa a las preferencias determinadas por el Usuario durante su visita al Sitio Web con el fin de reconocerlo como Usuario, y personalizar su experiencia y el uso del Sitio Web, y pueden también, por ejemplo, ayudar a identificar y resolver errores.
La información recabada a través de las cookies puede incluir la fecha y hora de visitas al Sitio Web, las páginas visionadas, el tiempo que ha estado en el Sitio Web y los sitios visitados justo antes y después del mismo. Sin embargo, ninguna cookie permite que esta misma pueda contactarse con el número de teléfono del Usuario o con cualquier otro medio de contacto personal. Ninguna cookie puede extraer información del disco duro del Usuario o robar información personal. La única manera de que la información privada del Usuario forme parte del archivo Cookie es que el usuario dé personalmente esa información al servidor.
Las cookies que permiten identificar a una persona se consideran datos personales. Por tanto, a las mismas les será de aplicación la Política de Privacidad anteriormente descrita. En este sentido, para la utilización de las mismas será necesario el consentimiento del Usuario. Este consentimiento será comunicado, en base a una elección auténtica, ofrecido mediante una decisión afirmativa y positiva, antes del tratamiento inicial, removible y documentado.
Cookies propias
Son aquellas cookies que son enviadas al ordenador o dispositivo del Usuario y gestionadas exclusivamente por Vertismed para el mejor funcionamiento del Sitio Web. La información que se recaba se emplea para mejorar la calidad del Sitio Web y su Contenido y su experiencia como Usuario. Estas cookies permiten reconocer al Usuario como visitante recurrente del Sitio Web y adaptar el contenido para ofrecerle contenidos que se ajusten a sus preferencias.
Cookies de terceros
Son cookies utilizadas y gestionadas por entidades externas que proporcionan a Vertismed servicios solicitados por este mismo para mejorar el Sitio Web y la experiencia del usuario al navegar en el Sitio Web. Los principales objetivos para los que se utilizan cookies de terceros son la obtención de estadísticas de accesos y analizar la información de la navegación, es decir, cómo interactúa el Usuario con el Sitio Web.
La información que se obtiene se refiere, por ejemplo, al número de páginas visitadas, el idioma, el lugar a la que la dirección IP desde el que accede el Usuario, el número de Usuarios que acceden, la frecuencia y reincidencia de las visitas, el tiempo de visita, el navegador que usan, el operador o tipo de dipositivo desde el que se realiza la visita. Esta información se utiliza para mejorar el Sitio Web, y detectar nuevas necesidades para ofrecer a los Usuarios un Contenido y/o servicio de óptima calidad. En todo caso, la información se recopila de forma anónima y se elaboran informes de tendencias del Sitio Web sin identificar a usuarios individuales.
Puede obtener más información sobre las cookies, la información sobre la privacidad, o consultar la descripción del tipo de cookies que se utiliza, sus principales características, periodo de expiración, etc. en el siguiente(s) enlace(s):
La(s) entidad(es) encargada(s) del suministro de cookies podrá(n) ceder esta información a terceros, siempre y cuando lo exija la ley o sea un tercero el que procese esta información para dichas entidades.
Cookies de redes sociales
Vertismed incorpora plugins de redes sociales, que permiten acceder a las mismas a partir del Sitio Web. Por esta razón, las cookies de redes sociales pueden almacenarse en el navegador del Usuario. Los titulares de dichas redes sociales disponen de sus propias políticas de protección de datos y de cookies, siendo ellos mismos, en cada caso, responsables de sus propios ficheros y de sus propias prácticas de privacidad. El Usuario debe referirse a las mismas para informarse acerca de dichas cookies y, en su caso, del tratamiento de sus datos personales. Únicamente a título informativo se indican a continuación los enlaces en los que se pueden consultar dichas políticas de privacidad y/o de cookies:
El Usuario puede deshabilitar, rechazar y eliminar las cookies —total o parcialmente— instaladas en su dispositivo mediante la configuración de su navegador (entre los que se encuentran, por ejemplo, Chrome, Firefox, Safari, Explorer). En este sentido, los procedimientos para rechazar y eliminar las cookies pueden diferir de un navegador de Internet a otro. En consecuencia, el Usuario debe acudir a las instrucciones facilitadas por el propio navegador de Internet que esté utilizando. En el supuesto de que rechace el uso de cookies —total o parcialmente— podrá seguir usando el Sitio Web, si bien podrá tener limitada la utilización de algunas de las prestaciones del mismo.




Lo sentimos, el formulario de comentarios está cerrado en este momento.