
La Enfermedad de Gaucher y su impacto
La primera descripción de esta enfermedad fue descrita por Pillipe Gaucher, en una tesis brillante cuya portada adjuntamos a continuación estudió a una paciente con esplenomegalia y que la describe como no asociada con leucemia, al estudiar las células consideró inicialmente que correspondían a malignas por su crecimiento, cuando se presentó otro caso fue conocida esta patología como enfermedad de Gaucher1.
Posteriormente se descubrió su patrón hereditario, pero se mantuvo el epónimo por consideración al médico que la descubrió. La enfermedad de Gaucher es un trastorno hereditario raro que permite la acumulación de sustancias grasosas en bazo, hígado, pulmones e inclusive en cerebro. Esta afección es causada por la herencia de tipo autosómico recesivo de una mutación del gen GBA que codifica la enzima ácida lisosomal glucocerebrosidasa, la glucocerebrosidasa es una enzima que es necesaria para el metabolismo de la glucosilceramida2. Este trastorno afecta el metabolismo de lípidos y si acumulación, cabe mencionar que no es la única enfermedad asociada a esta cadena metabólica, a continuación se adjunta un brillante esquema realizado por Alaa Abou Daher2.

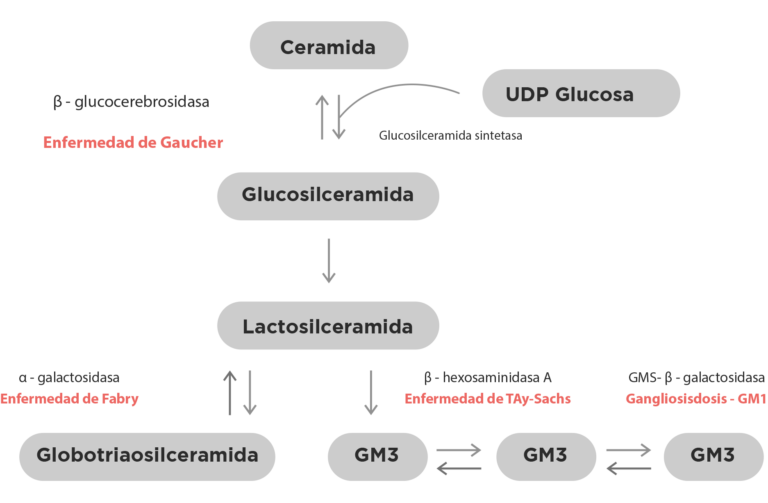
En lo referente a la clasificación de esta enfermedad, se han identificado tres fenotipos mayores, la distribución de estos que ha sido descrita a lo largo de los años no ha presentado mayor variación entre lo publicado en el año 20003 y se ha mantenido muy parecida en el 20174.

El fenotipo 1 es el que sufre la mayoría de los pacientes con esta enfermedad; es conocida como no neuropática, la dificultad de diagnóstico de este subgrupo es que se han encontrado casos con diagnóstico en séptima u octava década de la vida, dichos pacientes fueron diagnosticados como portadores de la enfermedad de Parkinson y luego de un estudio se demostró un fenotipo de Gaucher, esto muestra las dificultades del diagnóstico en estos pacientes5. Los otros subtipos son menos frecuentes, en el caso de la enfermedad tipo 2 tiene un pésimo pronóstico de corto plazo (2 a 3 años luego de nacer), este pronóstico está causado por el catastrófico cuadro neurológico, entre los síntomas que se mencionan están episodios de asfixia, apneas prolongadas entre otros6.
En el Ecuador
En prensa se recoge que en Ecuador hay unos 237 casos posibles tratados a lo largo del tiempo con esta enfermedad; sin embargo, por la falta de herramientas para estudio adecuado además de no tener un registro nacional hay una alta probabilidad de subdiagnóstico. Se ha revisado la literatura para indagar sobre casos y hasta el momento se han encontrado algunas publicaciones:
Tabla 1: Casos publicados con Gaucher en Ecuador.
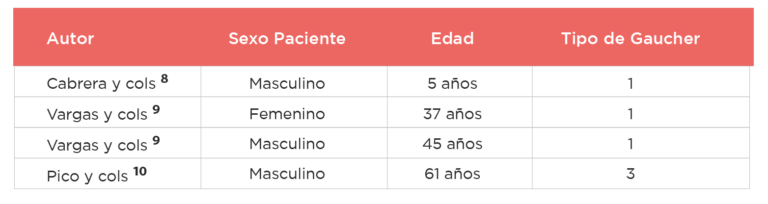
En el primer caso publicado por Cabrera y cols. se describe a un paciente de 5 años que desde el primer año de vida presentó distensión abdominal, esplenomegalia, hepatomegalia, equimosis espontáneas, anemia y trombocitopenia severas, ingresa por primera vez a hospitalización por un cuadro respiratorio y posteriormente pasa a estudio de esta patología encontrándose disminución de los niveles de glucocerebrosidasa y mutación homocigota en el exón 10 del gen GBA8.
En el segundo caso se describe a una paciente de 37 años que presentó desde los 14 años astenia y hematomas en miembros inferiores sin causa, este cuadro se mantiene estable, y se incrementa en el segundo embarazo en que se suma dolor óseo, distensión abdominal y sensación de plenitud gástrica, se encuentra en examen físico importante esplenomegalia y hepatomegalia en el estudio de laboratorio hay evidencia de Enfermedad de Gaucher9.
En el tercer caso se describe a un paciente de 45 años que tuvo una fractura en cuello de fémur a los 25 años, acude con astenia, plenitud gástrica y dolores óseos que no responden a tratamiento analgésico; en examen se destaca importante esplenomegalia y hepatomegalia. En ambos casos el inicio de tratamiento con tratamiento enzimático muestra buenos resultados9.
El cuarto caso corresponde a un paciente de 61 años con antecedente de Enfermedad de Gaucher tipo 3 sin terapia de reemplazo enzimática presente, esplenectomizado previamente que además presentaba cirrosis y múltiples fracturas, lo que llamó la atención a los médicos fue la presencia de múltiples nódulos en parénquima hepático, el diagnóstico histopatológico fue la presencia de gaucheroma hepático.
Los anteriores muestran varias formas de presentación de la enfermedad o de su evolución. Es importante recalcar los pocos casos que hacen en todas las enfermedades raras que los números de pacientes sean bajos, a continuación en la siguiente tabla presentaremos el número de pacientes reclutados en varios estudios fase 3, registrados en la página Clinical Trials que resumen a los estudios más importantes en el mundo:
En resumen es una enfermedad que en el país existen elementos para considerar la posibilidad de un alto subdiagnóstico, además de que los problemas en su evolución pueden enmascarar dicho diagnóstico hasta momentos tardíos del paciente.
BIBLIOGRAFIA:
1.- Gaucher PCE. De l’epithelioma primitive de las rate, hypertrophie idiopathique de la rate sans leucemie. {Academic Thesis} Paris, France; 1885.
2.- Messner, M., & Cabot, M. (2021). Glucosylceramide in Humans. Retrieved 16 July 2021, from https://pubmed.ncbi.nlm.nih.gov/20919653/
2.- Abou Daher, A., El Jalkh, T., Eid, A., Fornoni, A., Marples, B., & Zeidan, Y. (2021). Translational Aspects of Sphingolipid Metabolism in Renal Disorders. Retrieved 17 July 2021, from https://www.researchgate.net/publication/321322681_Translational_Aspects_of_Sphingolipid_Metabolism_in_Renal_Disorders
3.- Charrow, J., Andersson, H., Kaplan, P., Kolodny, E., Mistry, P., & Pastores, G. et al. (2000). The Gaucher Registry. Archives Of Internal Medicine, 160(18), 2835. doi: 10.1001/archinte.160.18.2835.
4.- Nalysnyk, L., Rotella, P., Simeone, J., Hamed, A., & Weinreb, N. (2016). Gaucher disease epidemiology and natural history: a comprehensive review of the literature. Hematology, 22(2), 65-73. doi: 10.1080/10245332.2016.1240391.
5.- Rosenbloom, B., Balwani, M., Bronstein, J., Kolodny, E., Sathe, S., & Gwosdow, A. et al. (2021). The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Retrieved 19 July 2021, from
6.- Mignot, C., Doummar, D., Maire, I., & De Villemeur, T. (2006). Type 2 Gaucher disease: 15 new cases and review of the literature. Brain And Development, 28(1), 39-48. doi: 10.1016/j.braindev.2005.04.005
7.- Hora, D. (2021). Enfermedad de Gaucher en Ecuador: 23 personas la padecen – La Hora. Retrieved 12 July 2021, from https://lahora.com.ec/noticia/1102198383/enfermedad-de-gaucher-en-ecuador-23-personas-la-padecen.
8.- Cabrera, A. (2019). Sociedad Ecuatoriana de Pediatría. Retrieved 12 July 2021, from https://docs.bvsalud.org/biblioref/2020/08/1116481/revista-pediatria-vol-20-no2-2019v1-min-53-58.pdf.
9.- Vargas, R. (2018). Retrieved 12 July 2021, from https://www.researchgate.net/publication/324866300_ENFERMEDAD_DE_GAUCHER_APORTACION_DE_2_CASOS_EVOLUCION_CON_TRATAMIENTO_ENZIMATICO_SUSTITUTIVO/link/5ae91c5caca2725dabb51c25/download.
10.- Poveda, M., Romero, E., Cárdenas, J., Tacuri, J., Grandes, I., & Balseca, V. (2019). Gaucheroma hepático. Un pseudotumor que puede confundir al radiólogo. Reporte de un caso. Revista Argentina De Radiología / Argentinian Journal Of Radiology, 84(01), 033-035. doi: 10.1055/s-0039-1693670.
Lo sentimos, el formulario de comentarios está cerrado en este momento.