

Cáncer Pediátrico
HABLEMOS DE CÁNCER INFANTIL: NEUROBLASTOMA, DIFERENTES ESPECTROS DENTRO DE UNA MISMA PATOLOGÍA
El cáncer infantil es considerado una de las principales causas de muerte en niños y adolescentes, entre los distintos tipos de esta enfermedad destacan las leucemias, linfomas y tumores en el sistema nervioso central, especialmente en este grupo poblacional. Aunque al pasar de los años la tasa de incidencia de esta enfermedad en la población ha aumentado, sigue siendo difícil realizar la prevención efectiva; una vez diagnosticado es de vital importancia la realización de un tratamiento oportuno.

Según la Clasificación Internacional del Cáncer Infantil (ICCC), los tumores neuroblásticos pertenecen a una familia de neoplasias clasificadas en el Grupo IV, de los cuales el ganglioneuroblastoma (GNB) y el neuroblastoma (NB) representan el 97%.[1] Los tumores neuroblásticos muestran un comportamiento biológico y clínico variado que va desde la regresión espontánea hasta la progresión, y pueden responder al tratamiento o volverse resistentes a él.
El neuroblastoma es el segundo tumor sólido más común en niños, comprende el 5% de todas las neoplasias infantiles. [4] Surge de las células ganglionares simpáticas primitivas y puede estar presente prenatalmente, en estos casos los niños pueden presentar una tumoración suprarrenal, abdominal, masa torácica, cervical o pélvica. El neuroblastoma comúnmente metastatiza a hueso cortical, médula ósea, piel, ganglios linfáticos y el hígado. [5]
La influencia de los agentes ambientales conocidos en la etiología de los tumores neuroblásticos, y en general de los tumores embrionarios, es muy baja, si alguna. Se han descrito casos de tumores neuroblásticos asociados a otros trastornos del desarrollo de la cresta neural como neurofibromatosis o enfermedad de Hirschprung, lo que sugiere un defecto genético común. En efecto, muy recientemente se han descrito mutaciones en el gen Phox2b en casos de tumores neuroblásticos con agregación familiar. [6]
La edad de presentación más común es entre los 18 y 22 meses, la mayoría de casos es diagnosticado antes de los 5 años de edad; la edad en el momento del diagnóstico es un indicador importante de curso clínico, siendo los bebés menores de 18 meses más propensos a padecer enfermedad que regresa espontáneamente o se trata con éxito solo con cirugía; a diferencia de niños mayores que tienen más probabilidades de tener tumores agresivos resistentes al tratamiento multimodal y terapias citotóxicas.
Los síntomas y signos de presentación clínica son claramente dependientes de la localización del tumor primario o las metástasis. Los tumores neuroblásticos se originan, en la mayor parte de los casos, en el retroperitoneo, bien en la glándula suprarrenal (44% de todos los casos), o bien en los ganglios paraespinales (22%). Otras regiones menos frecuentes son el mediastino posterior (15%), la pelvis (5%) o el cuello (<5%). La presentación más común es una masa abdominal indolora. Los síntomas, cuando están presentes, pueden estar relacionados al efecto de masa del tumor primario, secuelas de enfermedad metastásica o síndromes paraneoplásicos. Los síntomas sistémicos de pérdida de peso, fiebre y fatiga a menudo indican deterioro de la médula ósea, afectación y enfermedad avanzada. Los bebés también pueden presentar metástasis a modo de nódulos subcutáneos. El opsoclono-mioclono es una rara enfermedad inmunomediada manifestación paraneoplásica del neuroblastoma que a menudo persiste y puede estar asociada con ataxia; típicamente se asocia con tumores primarios pequeños y un resultado favorable [7].
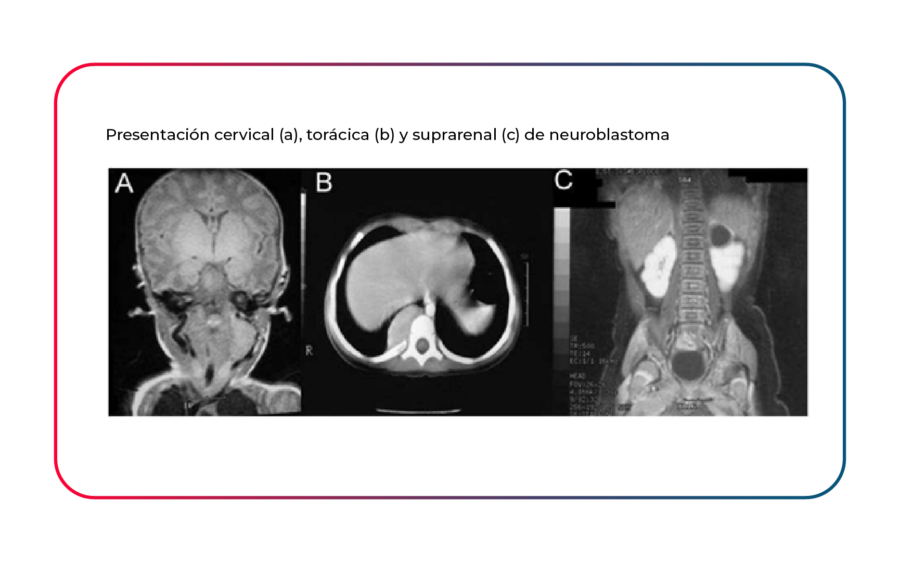

El origen de estos tumores explica algunas de las características clínicas de su comportamiento así como los lugares de presentación del tumor primario (médula suprarrenal, cadenas paravertebrales abdominal y mediastínica, cadena prevertebral pélvica); la captación de Meta Iodo Bencil Guanidina (MIBG), un radiofármaco análogo a los precursores catecolaminérgicos; y la secreción de catecolaminas o precursores adrenérgicos inmaduros.
El análisis de los datos mediante el sistema de clasificación desarrollado por el grupo internacional de riesgo del NB confirma el valor predictivo de la edad con un punto de corte a los 18 meses. La histología es otra variable importante para el pronóstico. Los factores biológicos, como el estado de MYCN y una deleción de 11q, se asocian con un pronóstico precario.[8] Según el estado de riesgo del paciente, el tratamiento incluye cirugía, quimioterapia, radioterapia, inmunoterapia y trasplante. En pacientes de alto riesgo, el tratamiento es intensivo y requiere un centro de atención terciaria.
Aunque la supervivencia en general ha mejorado, el 36% de los pacientes presenta enfermedad metastásica de alto riesgo que es difícil de curar[8]. Las tasas de supervivencia en enfermedad de alto riesgo se mantiene por debajo del 40% a pesar de la terapia multimodal que incluye quimioterapia mieloablativa, radioterapia, inmunoterapia y cirugía agresiva; de allí el advenimiento de las terapias biológicas con anticuerpos monoclonales dirigidos al disialogangliósido GD2 que han mejorado las tasas de supervivencia libre de evento así como la supervivencia global a 2 años. [9]
El neuroblastoma representa un amplio espectro de manifestaciones clínicas asociadas a comportamientos biológicos variables que requieren de una aproximación terapéutica diferencial, de allí la importancia en su oportuna identificación en los grupos de riesgo vulnerables porque solo se diagnostica lo que se reconoce.
Bibliografía
- Steliarova-Foucher E, Stiller C, Lacour B, Kaatsch P. International classification of childhood cancer, third edition. Cancer 2005;103:1457–1467.
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet Oncol 2007;369:2106–2120.
- Wood L, Lowis S. An update on neuroblastoma. Paediatr Child Health 2008;18:123–128
- Zhang Y, Huang D, Zhang W, Tang S, Han T, Zhu X, et al. Clinical characteristics of infant neuroblastoma and a summary of treatment outcome. Oncol Lett [Internet]. diciembre de 2016;12(6):5356- 62. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/ PMC5228312/
- Colon N, Chung D. Neuroblastoma. Adv Pediatr [Internet]. 2011;58(1):297-311. Disponible en: https://www.ncbi.nlm.nih.gov/ pmc/articles/PMC3668791/
- Trochet D, Bourdeaut F, Janoueix-Lerosey I, et al. Germline mutations of the paired-like homeobox 2B (Phox2B) gene in neuroblastoma. Am J Hum Genet. 2004 Apr;74(4):761-4
- Rudnick E, Khakoo Y, Antunes NL, Seeger RC, Brodeur GM, Shimada H, et al. Opsoclonus‐myoclonus‐ataxia syndrome in neuroblastoma: Clinical outcome and antineuronal antibodies—a report from the children’s cancer group study. Med Pediatr Oncol 2001;36:612–22. doi:10.1002/mpo.1138
- Cohn SL, Pearson ADJ, London WB, Monclair T, Ambros PF, Brodeur GM, Brisse J, Cecchetto G, Holmes K, Brisse H, Cecchetto G, Holmes K, Kaneko M, Matthay KK, Nuchtern JG, Von Schweinitz D, Simon The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J Clin Oncol 2009;27:289–297.
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 2010;363:1324–34. doi:10.1056/NEJMoa0911123
Lo sentimos, el formulario de comentarios está cerrado en este momento.