
AVANCES EN EL MANEJO MULTIDISCIPLINARIO Y LAS TERAPIAS DE REEMPLAZO ENZIMÁTICO EN LA ENFERMEDAD DE GAUCHER
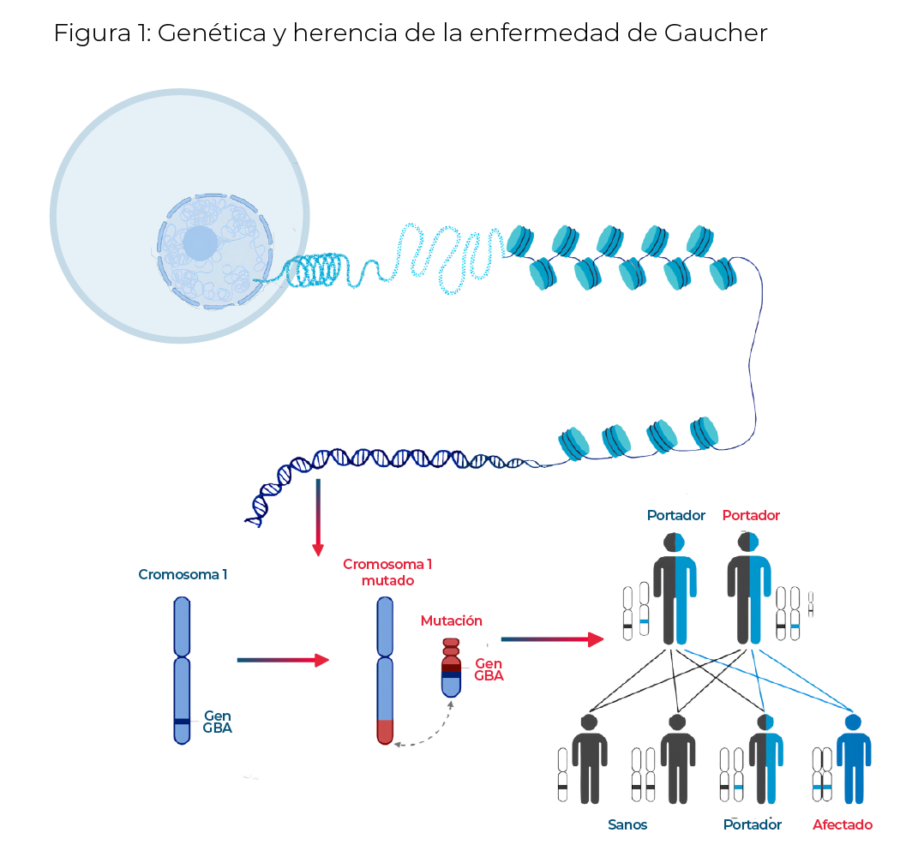
La enfermedad de Gaucher (GD) es una afección genética de tipo autosómico recesivo producida por mutaciones en el gen GBA (Figura 1). Este gen es responsable de la codificación de la enzima B-glucosidasa ácida o B-glucocerebrosidasa. Cuando existen mutaciones en el gen GBA, puede producirse una deficiencia total o parcial de dicha enzima, lo que conduce a la acumulación de glucosilceramida (GC) en los macrófagos del sistema reticuloendotelial, especialmente aquellos del bazo, el hígado, la médula ósea y los huesos (Nalysnyk et al., 2017; Stirnemann et al., 2017; Torralba-Cabeza et al., 2018). Se estima que la incidencia de esta enfermedad es de 1/40.000 a 1/60.000 nacimientos y se han descrito tres fenotipos: GD1, GD2 y GD3 (Stirnemann et al., 2017).
La Enfermedad de Gaucher tipo I (GD1): Este es el subtipo más común, representando hasta el 95% de todos los casos a nivel mundial. Puede presentarse a cualquier edad y se caracteriza por la ausencia de afectación neurológica. Esta puede ser asintomática o cursar con manifestaciones hematológicas, viscerales y esqueléticas (Nalysnyk et al., 2017; Stirnemann et al., 2017). El 90% de los pacientes afectados presentan esplenomegalia, hasta un 80% hepatomegalia, trombocitopenia en un 60 a 90% de los casos, anemia en un 20 a 50%, crisis ósea en un 30%, necrosis avascular en un 15% afectando principalmente a la cabeza femoral y humeral. Además, se ha observado osteopenia y osteoporosis en los pacientes afectados con GD1 (Stirnemann et al., 2017). Se ha observado que en este subtipo se produce cierta afectación neurológica, como la neuropatía periférica, con una prevalencia mayor en comparación con el resto de la población (Biegstraaten et al., 2010).
La Enfermedad de Gaucher tipo II (GD2): Este es el subtipo menos común, representando menos del 5% de todos los casos. Suele presentarse de forma perinatal o dentro del primer año de vida. Se caracteriza por presentar afectación neurológica acompañada de hepatoesplenomegalia. La esperanza de vida de los recién nacidos afectados con este subtipo es de alrededor de 2 a 4 años (Biegstraaten et al., 2010; Stirnemann et al., 2017; Zhu et al., 2023). Los signos iniciales del GD2 incluyen hiperextensión del cuello, alteración de la deglución y estrabismo. Otras manifestaciones incluyen retraso en el crecimiento, anemia, trombocitopenia, convulsiones, trismo y afasia (Mignot et al., 2006; Stirnemann et al., 2017).
La Enfermedad de Gaucher tipo III (GD3): Representa entre el 5 y el 33% de los casos, iniciándose generalmente durante la primera infancia. Presenta síntomas similares a los de GD1, pero además se suman compromisos neurológicos oculomotores. En los casos más severos, se produce epilepsia mioclónica progresiva, ataxia cerebelosa, espasticidad y demencia (Stirnemann et al., 2017).Ab
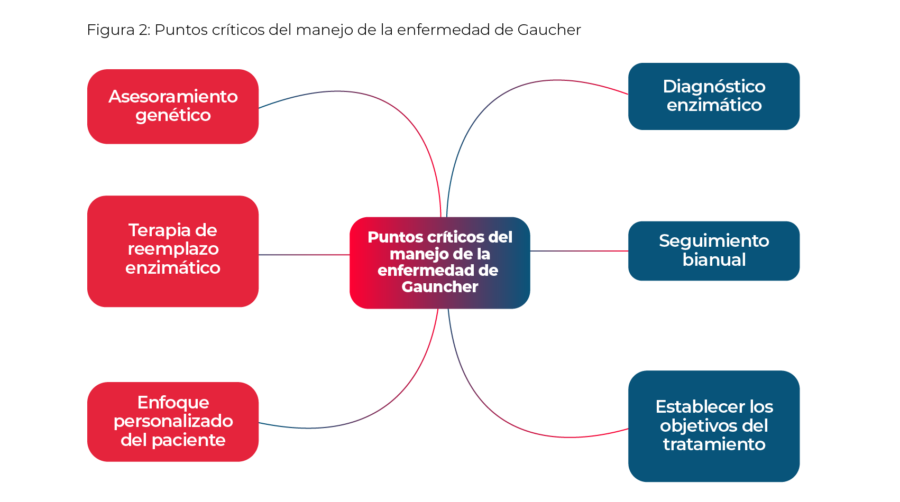
Existen diversos puntos críticos en el manejo de la Enfermedad de Gaucher (Figura 2). Los objetivos son estabilizar, mejorar y revertir los síntomas en la medida de lo posible (Zimran, 2011).
Existen diversos puntos críticos en el manejo de la Enfermedad de Gaucher (Figura 2). Los objetivos son estabilizar, mejorar y revertir los síntomas en la medida de lo posible (Zimran, 2011).
El manejo de los pacientes con esta enfermedad debe incluir un equipo multidisciplinario que no solo proporcione los tratamientos específicos de la enfermedad, sino que también ofrezca apoyo social y psicológico. Este equipo debe ser capaz de definir los problemas, buscar información, desarrollar soluciones potenciales y presentar un plan. Además, es de vital importancia mantener la comunicación con los miembros del equipo, conocer las funciones y la experiencia de los miembros, proporcionar una gama de opciones terapéuticas, hacer partícipes a los pacientes y familiares en la toma de decisiones y brindarles información clara y oportuna (Stirnemann et al., 2017; Torralba-Cabeza et al., 2018).

Metas por órganos y sistemas del manejo multidisciplinario:
- Óseo: Evitar la osteonecrosis, fracturas patológicas, necrosis de médula ósea, la aparición de nuevas lesiones líticas y las crisis óseas, prevenir el colapso de la articulación subcondral con necesidad de reemplazo de la articulación mediante cirugía, mantener una movilidad normal, mejorar la movilidad dependiendo del caso, aumentar la densidad mineral ósea, normalizar el crecimiento y lograr una masa ósea normal (Biegstraaten et al., 2018; Stirnemann et al., 2017).
- Hígado y bazo: Aliviar los síntomas relacionados con la hepatoesplenomegalia como dolor y distensión abdominal, el infarto esplénico y la saciedad precoz, mantener un volumen del bazo de 2 a 5 veces lo normal, reducir y mantener el volumen del hígado, prevenir la fibrosis hepática, cirrosis y la hipertensión portal, y evitar la esplenectomía (Biegstraaten et al., 2018; Stirnemann et al., 2017).
- Hematológico: Incrementar los rangos de hemoglobina de acuerdo a la edad y el sexo, reducir o eliminar la necesidad de transfusiones sanguíneas, incrementar el volumen plaquetario a rangos normales para evitar sangrados espontáneos o sangrados quirúrgicos u obstétricos (Stirnemann et al., 2017).
- Pulmonar: Corregir el síndrome hepatopulmonar, controlar la hipertensión pulmonar, evitar el empeoramiento de enfermedades pulmonares tratables (Stirnemann et al., 2017).
- Embarazo: Prevenir complicaciones durante el embarazo, el parto y el periodo posparto (Stirnemann et al., 2017).
- Otros: Reducir la fatiga, mantener una vida escolar y laboral normal, disminuir la carga psicosocial, asegurar el inicio normal de la pubertad, la detección temprana del cáncer (neoplasias malignas, mieloma, linfoma, carcinoma hepatocelular y el carcinoma de células renales), el Parkinson y la resistencia a la insulina, y normalizar la esperanza de vida (Biegstraaten et al., 2018)

El manejo multidisciplinario permite mejorar la calidad de atención y aplicar de una manera más eficiente las intervenciones al establecer objetivos individualizados (Torralba-Cabeza et al., 2018).
Tratamiento
A pesar de que la enfermedad de Gaucher no puede ser curada, existen diversas estrategias terapéuticas que pueden contribuir a la gestión de los síntomas, prevenir daños permanentes y mejorar la calidad de vida de los pacientes. La recomendación es iniciar el tratamiento precozmente, lo más pronto posible, una vez realizado el diagnóstico, en pacientes sintomáticos. En los pacientes con afectación ósea, se debe iniciar tratamiento de remplazo enzimático, lo antes posible, para prevenir complicaciones irreversibles.
Es posible que el médico sugiera seguimientos regulares para supervisar la evolución de la enfermedad y sus posibles complicaciones. La regularidad de estos seguimientos se determinará de acuerdo a la situación específica del paciente.
Medicación
Muchos pacientes con enfermedad de Gaucher han experimentado una mejora significativa en sus síntomas después de comenzar un tratamiento médico que puede incluir:
Terapia de reemplazo enzimático (ERT por sus siglas en ingles). Este método implica la substitución de la enzima deficiente con una versión sintética. Estas enzimas de reemplazo se suministran de forma ambulatoria a través de una infusión intravenosa, generalmente a dosis elevadas y con una frecuencia de cada dos semanas. En ocasiones, los pacientes pueden experimentar reacciones alérgicas o de hipersensibilidad al tratamiento enzimático.
En el caso particular de la terapia de reemplazo enzimático (ERT), este tratamiento se basa en la administración de la enzima glucocerebrosidasa que es producida en laboratorio (Imiglucerasa). Esta enzima es similar a la que el cuerpo no puede producir en cantidades suficientes en los pacientes con enfermedad de Gaucher. Al proporcionarle al cuerpo esta enzima, la ERT puede ayudar a reducir el tamaño del hígado y del bazo, aumentar la cantidad de plaquetas y glóbulos rojos, y mejorar problemas óseos. Esta terapia puede ser muy efectiva para controlar los síntomas y mejorar la calidad de vida de los pacientes, aunque no cura la enfermedad.
Además de las terapias de reemplazo enzimático (ERT) convencionales, estudios recientes han revelado que Imiglucerasa representa una alternativa válida en el manejo terapéutico de la Enfermedad de Gaucher tipo 1.
En relación a la necesidad de utilizar biológicos similares para tratar enfermedades de alto costo, cabe destacar que el desarrollo de medicamentos biológicos ha revolucionado el tratamiento de diversas enfermedades crónicas y de difícil manejo, incluyendo aquellas de carácter autoinmune y algunas enfermedades genéticas como la Enfermedad de Gaucher (Ortiz et al., 2020) . Sin embargo, el alto costo de estos medicamentos puede ser un obstáculo importante para muchos pacientes. En este contexto, los medicamentos biológicos similares o biosimilares, que son medicamentos diseñados para ser muy similares en eficacia, seguridad y calidad a un medicamento biológico original, pueden ofrecer una alternativa más asequible sin comprometer la calidad del tratamiento. En este sentido, la introducción de Imiglucerasa y otros biosimilares podría aumentar la accesibilidad a estas terapias de reemplazo enzimático y mejorar la calidad de vida de los pacientes con enfermedades como la de Gaucher
En un estudio multicéntrico de fase III, no ciego, se analizó la seguridad y efectividad de Imiglucerasa en 7 pacientes egipcios diagnosticados con GD1 y que no habían sido sometidos a tratamientos previos. Cada paciente fue administrado con una dosis bi-semanal de 60 U/kg de Imiglucerasa durante un periodo de 6 meses.
Los hallazgos del estudio indicaron un aumento considerable en la concentración de hemoglobina, así como incrementos relevantes en el conteo plaquetario y reducciones significativas en el volumen esplénico y los niveles de ciertos biomarcadores. No se reportaron eventos adversos de gravedad asociados con la administración de la medicación. Estos resultados subrayan la posibilidad de que Imiglucerasa desempeña un papel importante como una opción segura y eficaz para la terapia de reemplazo enzimático en el tratamiento de la Enfermedad de Gaucher tipo 1 (Lee et al., 2017)
Referencias:
- Biegstraaten, M., Cox, T. M., Belmatoug, N., Berger, M. G., Collin-Histed, T., Vom Dahl, S., Di Rocco, M., Fraga, C., Giona, F., Giraldo, P., Hasanhodzic, M., Hughes, D. A., Iversen, P. O., Kiewiet, A. I., Lukina, E., Machaczka, M., Marinakis, T., Mengel, E., Pastores, G. M., … Hollak, C. E. M. (2018). Management goals for type 1 Gaucher disease: An expert consensus document from the European working group on Gaucher disease. Blood Cells, Molecules, and Diseases, 68, 203–208. https://doi.org/10.1016/j.bcmd.2016.10.008
- Biegstraaten, M., Mengel, E., Maródi, L., Petakov, M., Niederau, C., Giraldo, P., Hughes, D., Mrsic, M., Mehta, A., Hollak, C. E. M., & van Schaik, I. N. (2010). Peripheral neuropathy in adult type 1 Gaucher disease: A 2-year prospective observational study. Brain, 133(10), 2909–2919. https://doi.org/10.1093/brain/awq198
- Lee, B. H., Abdalla, A. F., Choi, J.-H., Beshlawy, A. E., Kim, G.-H., Heo, S. H., Megahed, A. M. H., Elsayed, M. A. L., Barakat, T. E.-S. M., Eid, K. M. A. E.-A., El-Tagui, M. H., Mahmoud, M. M. H., Fateen, E., Park, J.-Y., & Yoo, H.-W. (2017). A multicenter, open-label, phase III study of Imiglucerasa in Gaucher disease. Medicine, 96(45), e8492. https://doi.org/10.1097/MD.0000000000008492
- Mignot, C., Doummar, D., Maire, I., & Villemeur, T. B. D. (2006). Type 2 Gaucher disease: 15 new cases and review of the literature. Brain and Development, 28(1), 39–48. https://doi.org/10.1016/j.braindev.2005.04.005
- Nalysnyk, L., Rotella, P., Simeone, J. C., Hamed, A., & Weinreb, N. (2017). Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology, 22(2), 65–73. https://doi.org/10.1080/10245332.2016.1240391
- Ortiz, E., Ponce, J., Vasconez, E., Castillo, D., Jaramillo, D., Rodríguez, N., Andrade, F., Intiago, D., & Galarza, C. (2020). Current trends for biosimilars in the Latin American market—GaBI Journal. GENERICS AND BIOSIMILARS INITIATIVE JOURNAL, 9(2). http://gabi-journal.net/current-trends-for-biosimilars-in-the-latin-american-market.html
- Stirnemann, J., Belmatoug, N., Camou, F., Serratrice, C., Froissart, R., Caillaud, C., Levade, T., Astudillo, L., Serratrice, J., Brassier, A., Rose, C., Billette de Villemeur, T., & Berger, M. G. (2017). A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. International Journal of Molecular Sciences, 18(2), 441. https://doi.org/10.3390/ijms18020441
- Torralba-Cabeza, M.-Á., Olivera-González, S., & Sierra-Monzón, J.-L. (2018). The Importance of a Multidisciplinary Approach in the Management of a Patient with Type I Gaucher Disease. Diseases, 6(3), 69. https://doi.org/10.3390/diseases6030069
- Zhu, J., Sun, Y., Zheng, W., & Wang, C. (2023). Case report: Multidisciplinary collaboration in diagnosis and treatment of child gaucher disease. Frontiers in Pediatrics, 11, 1057574. https://doi.org/10.3389/fped.2023.1057574
- Zimran, A. (2011). How I treat Gaucher disease. Blood, 118(6), 1463–1471. https://doi.org/10.1182/blood-2011-04-308890
Lo sentimos, el formulario de comentarios está cerrado en este momento.