
Enfermedad de Gaucher
La enfermedad de Gaucher es una afección congénita de herencia autosómica recesiva que afecta al metabolismo. Esta enfermedad es causada por mutaciones en el gen GBA1, ubicado en el cromosoma 1, que codifica una enzima encargada del catabolismo de los glucocerebrósidos. El gen GBA1 presenta diversos tipos de mutaciones, que incluyen inserciones, deleciones y alelos complejos. Las mutaciones más comunes son 84GG, IVS2+1, p.N370S y L444P (Pastores y Hughes, 1993; Stirnemann et al., 2017; Stone, Basit y Master, 2023). La enfermedad es más prevalente en la población judía Ashkenazi, con una frecuencia de portadores del 6 %, en comparación con el 0.7 % al 0.8 % en la población no judía. La incidencia de la enfermedad varía según la fuente, y se encuentra en un rango que va de 0.39 por 100,000 hasta 5.80 por 100,000 (Nalysnyk et al., 2017; Stone et al., 2023).
Síntomas:
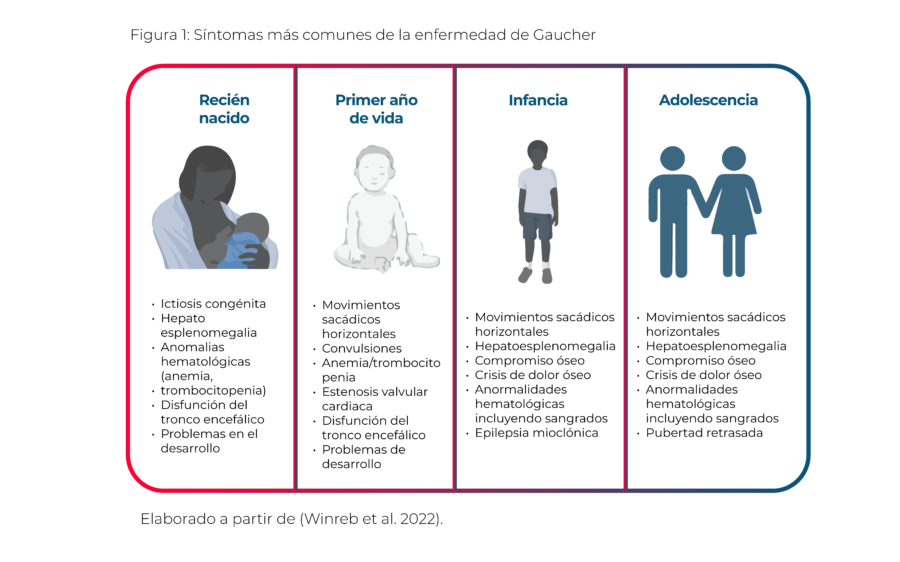
La enfermedad de Gaucher presenta numerosos síntomas, siendo los más destacados la esplenomegalia, hepatomegalia, dolor óseo, anemia, trombocitopenia, movimientos sacádicos horizontales lentos con visión intacta, deterioro del desarrollo motor primario y epilepsia mioclónica (Weinreb et al., 2022).
Clasificación
Tipo 1: Esta es la forma más común de la enfermedad de Gaucher, representando el 90 % de los casos en Europa y el 95 % en América del Norte. La edad media de diagnóstico oscila entre 10 y 20 años. La presentación clínica es variable, pudiendo ser asintomática a lo largo de la vida o manifestar síntomas en la infancia. Los síntomas clínicos principales incluyen esplenomegalia, trombocitopenia, infiltración de médula ósea, hepatomegalia, fatiga, anemia, crisis óseas dolorosas agudas y necrosis avascular. No hay deterioro neurológico en este tipo, aunque el sistema nervioso puede verse afectado secundariamente debido a problemas hematológicos y esqueléticos graves (ALAEI et al., 2019; Stirnemann et al., 2017).
Tipo 2: Representa aproximadamente el 1 % de los casos, aunque algunas cohortes informan hasta un 20 %. La incidencia estimada es de 1 en 150,000. Se caracteriza por un deterioro neurológico grave y progresivo que comienza entre los 3 y 6 meses de edad. Los pacientes afectados suelen fallecer entre los 9 meses y los dos años (ALAEI et al., 2019; Bohra and Nair, 2011; Stirnemann et al., 2017).
Tipo 3: Conocida como forma neurológica juvenil, representa el 5 % de todos los casos. Este tipo se divide en 3 subtipos: 3A, 3B y 3C, cada uno con características neurológicas y viscerales específicas (Bohra and Nair, 2011).
Perinatal-Letal: Esta es la forma más grave y tiene una prevalencia inferior al 1 %. Se caracteriza por la presencia de hidropesía fetal no inmunitaria y anomalías cutáneas neonatales como ictiosiforme-colodión. Además, la triada de hidropesía, ictiosis y acinesia fetal se ha asociado con mutaciones graves (ALAEI et al., 2019).
Diagnóstico
El diagnóstico de la enfermedad de Gaucher debe confirmarse mediante la determinación de una actividad deficiente de la enzima GCasa en leucocitos totales, células mononucleares o fibroblastos cultivados. También se pueden utilizar gotas de sangre seca para análisis enzimáticos, siendo la citometría de flujo de monocitos sanguíneos el método más preciso. El diagnóstico prenatal es posible y se puede realizar mediante análisis genéticos. Las muestras variarán según la edad gestacional; en las semanas 10 a 12 de amenorrea se emplean muestras de vellosidades coriónicas, y a las 16 semanas se utilizan células del líquido amniótico (Stirnemann et al., 2017).
Tratamiento.
El tratamiento estándar para la enfermedad de Gaucher es la terapia de reemplazo enzimático (TRE), que implica la administración regular de la enzima glucocerebrosidasa recombinante. Esta terapia ayuda a aliviar muchos de los síntomas más debilitantes y a mejorar la calidad de vida de los pacientes. La imiglucerasa para inyección es un biosimilar que ha sido aprobado para el uso como terapia de reemplazo enzimático a largo plazo en pacientes con un diagnóstico confirmado de enfermedad de Gaucher tipo 1. Al ser un biosimilar, la imiglucerasa ofrece una alternativa más rentable a los tratamientos de marca, manteniendo un perfil de eficacia y seguridad similar. Es especialmente indicado para mejorar parámetros hemáticos y reducir la esplenomegalia y la hepatomegalia. Es esencial que el tratamiento con imiglucerasa o cualquier otro producto de TRE se realice bajo la supervisión de un médico con experiencia en el manejo de trastornos metabólicos hereditarios.
Con la introducción de biosimilares como la imiglucerasa, la gestión de la enfermedad de Gaucher se vuelve más accesible para un mayor número de pacientes, lo que es un avance significativo en el tratamiento de esta enfermedad rara pero debilitante.
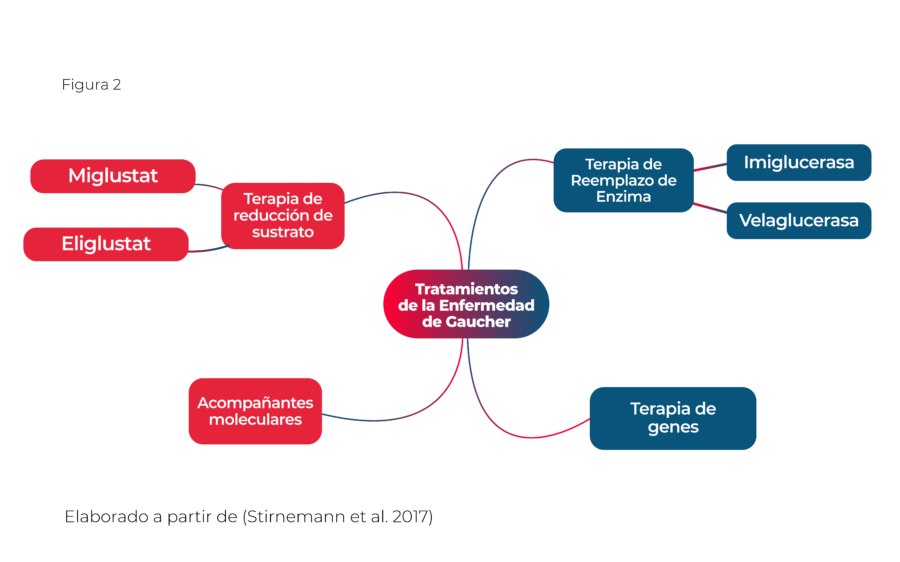
En la Figura 2 se mencionan los tratamientos actuales para la enfermedad de Gaucher
Conclusión.
Aunque la enfermedad de Gaucher es una condición rara, su conocimiento y entendimiento son fundamentales para la medicina y la salud pública. La variabilidad en su presentación clínica, que puede ir desde síntomas casi imperceptibles hasta manifestaciones severamente debilitantes, subraya la importancia de su diagnóstico temprano y preciso. La inclusión de terapias de reemplazo enzimático y la introducción de biosimilares como la imiglucerasa han revolucionado su tratamiento, mejorando sustancialmente la calidad de vida de los pacientes afectados. La enfermedad también sirve como un modelo para entender los mecanismos patofisiológicos subyacentes en una variedad de trastornos metabólicos y enfermedades lisosomales. Por tanto, aunque rara, la enfermedad de Gaucher es un eslabón crítico en la cadena de la investigación biomédica y clínica que puede ofrecer insights más amplios sobre enfermedades más comunes. Es imperativo que se siga investigando y divulgando información sobre este trastorno para fomentar diagnósticos más precisos, tratamientos más eficaces y, en última instancia, una mejor calidad de vida para los pacientes.
Bibliografía
- ALAEI, Mohammad Reza, Aydin TABRIZI, Narjes JAFARI, and Hadi MOZAFARI. 2019. “Gaucher Disease: New Expanded Classification Emphasizing Neurological Features.” Iranian Journal of Child Neurology 13(1):7–24.
- Bohra, Vijay, and Velu Nair. 2011. “Gaucher’s Disease.” Indian Journal of Endocrinology and Metabolism 15(3):182–86. doi: 10.4103/2230-8210.83402.
- Nalysnyk, Luba, Philip Rotella, Jason C. Simeone, Alaa Hamed, and Neal Weinreb. 2017. “Gaucher Disease Epidemiology and Natural History: A Comprehensive Review of the Literature.” Hematology 22(2):65–73. doi: 10.1080/10245332.2016.1240391.
- Pastores, Gregory M., and Derralynn A. Hughes. 1993. “Gaucher Disease.” in GeneReviews®, edited by M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Gripp, and A. Amemiya. Seattle (WA): University of Washington, Seattle.
- Stirnemann, Jérôme, Nadia Belmatoug, Fabrice Camou, Christine Serratrice, Roseline Froissart, Catherine Caillaud, Thierry Levade, Leonardo Astudillo, Jacques Serratrice, Anaïs Brassier, Christian Rose, Thierry Billette de Villemeur, and Marc G. Berger. 2017. “A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments.” International Journal of Molecular Sciences 18(2):441. doi: 10.3390/ijms18020441.
- Stone, William L., Hajira Basit, and Samip R. Master. 2023. “Gaucher Disease.” in StatPearls. Treasure Island (FL): StatPearls Publishing.
- Weinreb, Neal J., Ozlem Goker-Alpan, Priya S. Kishnani, Nicola Longo, T. Andrew Burrow, John A. Bernat, Punita Gupta, Nadene Henderson, Helio Pedro, Carlos E. Prada, Divya Vats, Ravi R. Pathak, Ekaterina Wright, and Can Ficicioglu. 2022. “The Diagnosis and Management of Gaucher Disease in Pediatric Patients: Where Do We Go from Here?” Molecular Genetics and Metabolism 136(1):4–21. doi: 10.1016/j.ymgme.2022.03.001.
Lo sentimos, el formulario de comentarios está cerrado en este momento.